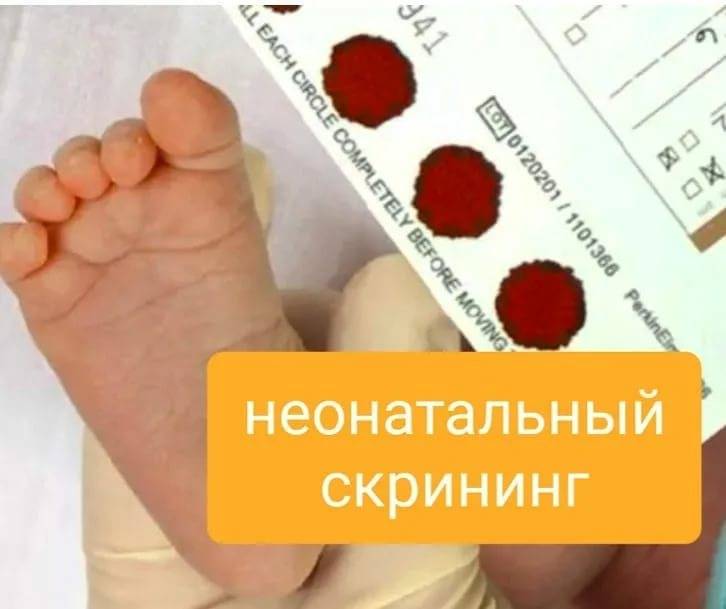
|
Стремительное развитие генетики позволило выяснить, что наследственная и врожденная патология является одной из основных причин как детской, так и взрослой, инвалидности и смертности. |
90% наследственных синдромов клинически проявляются в детском возрасте, а примерно половина – уже на первом году жизни. Для многих из них существует специфическое лечение. Поэтому раннее выявление врожденных и (или) наследственных заболеваний позволяет своевременно назначить адекватную лекарственную терапию или, соответствующую диету, что приводит к полной компенсации заболевания и, как следствие, отсутствию нарушений функций организма, в связи с чем детям не требуется установления инвалидности.
В Краснодарском крае с 1987 г. начато проведение неонатального скрининга на фенилкетонурию, с 1994 г. - на врождённый гипотиреоз.
Начиная с 2006 г. в Краснодарском крае, как и в целом по РФ, проводится неонатальный скрининг с целью раннего выявления 5 наследственных болезней обмена: фенилкетонурии, врождённого гипотиреоза, мусковисцидоза, адреногенитального синдрома, галактоземиии. В медицинских организациях службы родовспоможения у новорожденных берется кровь из пяточки и в виде сухих пятен на специальном тест-бланке доставляется в лабораторию медикогенетической консультации для проведения исследования.
За 17 лет проведения неонатального скрининга в Краснодарском крае обследованы более 1 млн новорожденных детей, выявлены 714 детей с наследственными болезнями обмена, из них фенилкетонурия – 145, врождённый гипотиреоз – 325, адреногенитальный синдром – 108, мусковицидоз – 106, галактоземияя – 30.
С 1 января 2023 г. стартовал проект по проведению расширенного неонатального скрининга. Теперь обследование новорожденных проводится на 36 заболеваний, из них 5, которые проводились ранее, и на 31 новое заболевание, в их числе спинальная мышечная атрофия, первичные иммунодефициты и наследственные нарушения обмена.
Часто задаваемые вопросы
Зачем новорожденному ребенку нужно пройти обследование?
Цель неонатального скрининга – выявить редкие, но тяжелые заболевания еще до развития их симптомов и вовремя начать лечение.
Заболевания, на которые проводится обследование, очень редкие, и риск их наличия у ребенка крайне низкий. Однако в соответствии с генетическими законами наследования этих болезней отсутствие случаев заболевания у родственников не исключает риска для ребенка.
Как и когда будет взят анализ у ребенка?
Кровь из пяточки у ребенка должны взять на специальный тест-бланк при доношенном сроке в первые 24-48 часов жизни в родильном доме. У недоношенных детей анализ должны взять на 7-е сутки жизни.
Если у ребенка не успели взять кровь?
Неонатальный скрининг проводится в определенные сроки для того, чтобы своевременно выявить заболевание и начать лечение. Поэтому не надо откладывать прохождение скрининга на более позднее время. Если же по разным причинам не удалось пройти обследование в родильном доме (ранняя выписка, роды на дому), провести его как можно скорее, обратившись в поликлинику по месту проживания. Не следует отказываться от проведения обследования, так как симптомы некоторых заболеваний не проявляются сразу после рождения, могут проявиться позднее.
Как узнать, был ли обследован ребенок?
При взятии крови для обследования в роддоме ставится отметка о прохождении скрининга в выписке из истории развития ребенка и обменной карте. Если кровь была взята в поликлинике по месту жительства – отметка ставится в амбулаторной карточке.
На какие заболевания проводится обследование?
Это фенилкетонурия, врожденный гипотиреоз, адреногенитальный синдром, муковисцидоз, галактоземия, спинальная мышечная атрофия, первичный иммунодефицит, наследственные болезни обмена.
Как узнают о результатах обследования?
Отсутствие вызова на дополнительное обследование будет означать нормальные результаты анализа по всем обследуемым заболеваниям.
Если у ребенка положительный результат анализа?
В случае подозрения на какое-либо заболевание родители ребенка в течение 24 часов получают вызов на дальнейшее обследование в медикогенетической консультации ГБУЗ «Научно-исследовательский институт им.
проф. С.В. Очаповского» министерства здравоохранения Краснодарского края.
В этом случае следует помнить, что первоначальный положительный результат не всегда означает наличие заболевания. Однако не стоит откладывать прохождение дополнительного обследования.
ЧТО ТАКОЕ ФЕНИЛКЕТОНУРИЯ?
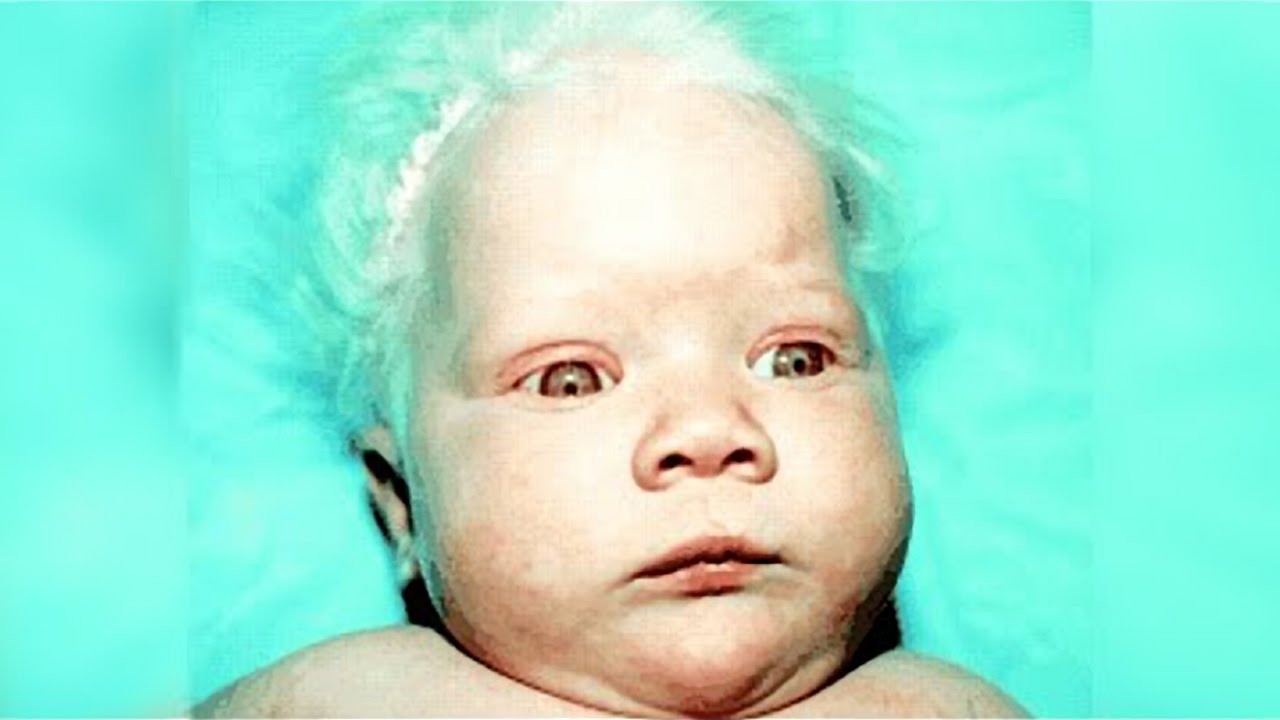
Фенилкетонурия– наследственное заболевание, которое встречается с частотой 1 на 5000 – 7000 новорожденных. Развитие этого заболевания связано с повреждением (мутацией) особого гена. Больной ребенок рождается, если он получает два мутантных гена (по одному от каждого из родителей). Родители больного ребенка всегда здоровы, но являются носителями патологического гена. Частота носительства мутаций, вызывающих фенилкетонурию, среди населения 1 на 40 человек. До рождения больного ребенка родители не знают о своем носительстве. Риск рождения больного ребенка в браке двоих таких носителей составляет 25% при каждой беременности. Все остальные родственники чаще всего здоровы.
Как проявляется заболевание?
Вследствие мутации гена страдает функция фермента фенилаланингидроксилазы, который обеспечивает пути обмена аминокислоты фенилаланина. Фенилаланин поступает в организм ребенка только с пищей. У больного фенилкетонурией постепенно повышается уровень ФА в крови. Высокий уровень фенилаланина является токсичным прежде всего для нервной системы. При отсутствии лечения после 3-6 месяцев жизни появляются клинические признаки болезни: отставание в нервно-психическом развитии, судороги, дерматиты, повышенная потливость со специфическим запахом пота и мочи, осветление кожи и волос. Впоследствии у детей, не получавших лечение, развивается глубокая умственная отсталость.
Рано начатое лечение позволяет предотвратить развитие всех этих клинических проявлений. Дети развиваются полноценно и внешне не отличаются от здоровых людей. Поэтому важно начать лечение как можно раньше!
Какое существует лечение заболевания?
Лечение заключается в назначении диетотерапии с ограничением белка, поступающего с обычной пищей, т.е. в диете исключаются все высокобелковые продукты. Рацион больных фенилкетонурией состоит из лечебной смеси аминокислот без фенилаланина, овощных и фруктовый блюд, малобелковых продуктов на крахмальной основе.
Кто будет наблюдать ребенка в случае подтверждения диагноза?
Основным врачом, наблюдающим больного ребенка, должны быть врачдиетолог и врач-генетик, имеющие опыт в расчете диеты ребенку, которые научат родителей расчету диеты для ребенка. В дальнейшем родители будут регулярно посещать врача для контроля за лечением, контроля за уровнем фенилаланина и расчета диеты. Лечебные смеси для ребенка родители будут получать по рецептам в аптеке по месту жительства.
ЧТО ТАКОЕ ВРОЖДЕННЫЙ ГИПОТИРЕОЗ?
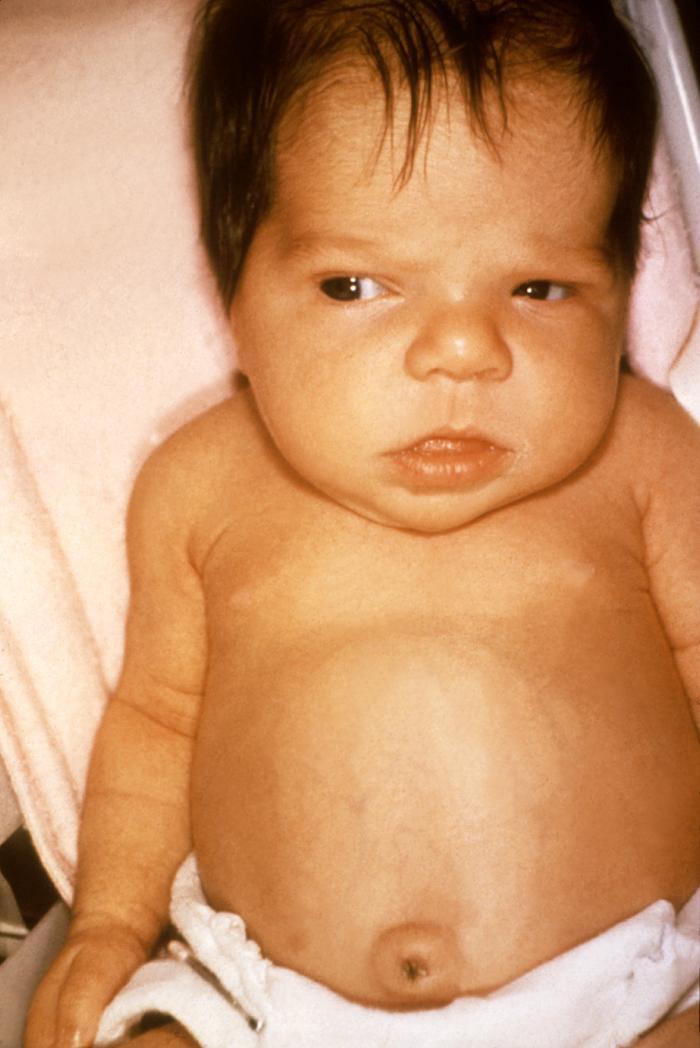
Врожденный гипотиреоз– врожденное заболевание, которое встречается у одного из 2 000 – 3 000 новорожденных. Его причины разнообразны, но чаще всего оно обусловлено врожденным пороком развития щитовидной железы. В этом случае чаще всего все родственники в семье здоровы, а риск повторного рождения ребенка с врожденным гипотиреозом не превышает 2-3%. Редко заболевание имеет наследственный характер и связано с получением ребенком двух поврежденных генов (по одному от каждого из родителей), что приводит к нарушению продукции гормонов в щитовидной железе. В таком браке родители абсолютно здоровы, но при каждой беременности существует риск рождения ребенка с врожденным гипотиреозом, равный 25%.
Как проявляется заболевание?
Гормоны щитовидной железы очень важны для нормальной работы всех органов, физического и психического развития ребенка. При тяжелой форме заболевания уже с рождения или первых недель жизни родители или врачпедиатр могут обратить внимание на вялость ребенка, отечность, затянувшуюся желтуху, хриплый голос, запоры, плохо заживающую пупочную ранку, сухость кожи, увеличенный язык. Менее тяжелые формы могут не иметь никаких симптомов заболевания в течение первых месяцев жизни. В дальнейшем при отсутствии лечения развивается задержка физического и психического развития ребенка.
Какое существует лечение заболевания?
Для лечения используются препарат L-тироксин, который восполняет дефицит собственных гормонов. Доза препарата подбирается индивидуально врачом-эндокринологом в зависимости от возраста, веса ребенка и степени тяжести заболевания.
Кто будет наблюдать ребенка в случае подтверждения диагноза?
Основным врачом, наблюдающим больного ребенка, будет эндокринолог. Частота посещения этого врача будет определяться индивидуально. В целом, на первом году жизни потребуются более частые визиты для оптимального подбора дозы препарата, так как вес малыша будет быстро меняться. Для того чтобы оценить эффективность и правильность лечения, будут необходимы регулярные анализы крови на гормоны щитовидной железы.
Какие отклонения в здоровье могут быть у ребенка в будущем?
В случае раннего начала лечения, правильного приема рекомендуемой терапии и регулярного наблюдения эндокринологом ребенок будет хорошо себя чувствовать, иметь соответствующее возрасту физическое и психическое развитие.
Может ли заболевание излечиться?
При небольшом отклонении от нормы гормонов щитовидной железы возможна самостоятельная нормализация функции щитовидной железы в течение первого года жизни. В этом случае лечение назначается далеко не всегда, но ребенку все же требуется наблюдение врачом-эндокринологом. Такое состояние называется «транзиторный гипотиреоз», а его причинами могут быть недоношенность ребенка, патология щитовидной железы или дефицит йода у мамы. В случае значительного отклонения гормонов щитовидной железы от нормы заболевание чаще всего не проходит со временем, так как обычно связано с неправильно сформированной щитовидной железой или генетически обусловленным нарушением продукции ее гормонов. В этом случае пациенту требуется пожизненный прием L-тироксина.
ЧТО ТАКОЕ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ?
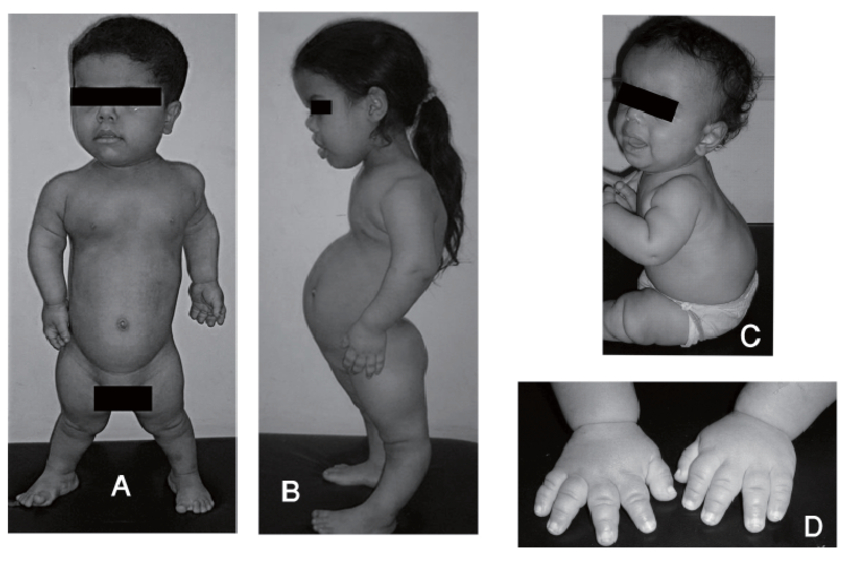
Адреногенитальный синдром – редкое наследственное заболевание, которое встречается у одного из 5 000 – 9 000 новорожденных. Оно связано с повреждением (мутацией) особого гена. Для того чтобы заболевание проявилось, ребенок должен получить две мутации (по одной от каждого из родителей). Родители больного ребенка всегда здоровы, но являются носителями патологического гена. До рождения больного ребенка родители не знают о своем носительстве. Риск рождения больного ребенка в браке двоих таких носителей составляет 25% при каждой беременности. Все остальные родственники чаще всего здоровы.
Как проявляется заболевание?
Мутации в указанном гене приводят к нарушению образования некоторых гормонов в особых железах организма – надпочечниках. Дефицит этих гормонов может по-разному проявляться у ребенка. У девочек при рождении отмечается неправильное строение наружных половых органов, что в некоторых случаях может привести к ошибочному определению пола ребенка. В первые недели жизни у новорожденного может развиться угрожающее жизни состояние, связанное с потерей солей. Оно проявляется обезвоживанием, рвотой, потерей массы тела и, при отсутствии соответствующей медицинской помощи или неправильно установленном диагнозе, может привести к смерти ребенка.
Заболевание может протекать и в достаточно легкой форме, когда у новорожденного ребенка отсутствуют какие-либо его проявления. Однако в будущем эта форма заболевания может привести к задержке роста, нарушению полового развития и репродуктивной функции.
Какое существует лечение заболевания?
Для лечения используются препараты гормонов надпочечников, которые восполняют дефицит собственных гормонов. Препараты подбираются индивидуально врачом-эндокринологом в зависимости от возраста, веса ребенка, степени тяжести и формы его заболевания. Девочкам, имеющим неправильное строение наружных половых органов, проводится ряд пластических операций.
Кто будет наблюдать ребенка в случае подтверждения диагноза?
Основным врачом, наблюдающим больного ребенка, будет врачэндокринолог. Частота посещения этого врача будет определяться индивидуально. Для того чтобы оценить эффективность и правильность лечения, будут необходимы регулярные анализы крови на гормоны.
Какие отклонения в здоровье будут у ребенка в будущем?
Правильно подобранное и соблюдаемое лечение позволит избежать кризов, связанных с потерей солей. Девочкам, имеющим нарушение строения половых органов при рождении, возможно потребуется хирургическая коррекция. Ребенок может иметь небольшую задержку роста и окончательный рост ниже среднего. В ряде случаев отмечается более раннее, чем в среднем начало полового созревания. Девушкам и женщинам (в том числе с легкой формой заболевания) потребуется наблюдение врача-акушера-гинеколога, особенно в период полового созревания и в репродуктивном возрасте.
ЧТО ТАКОЕ МУКОВИСЦИДОЗ?
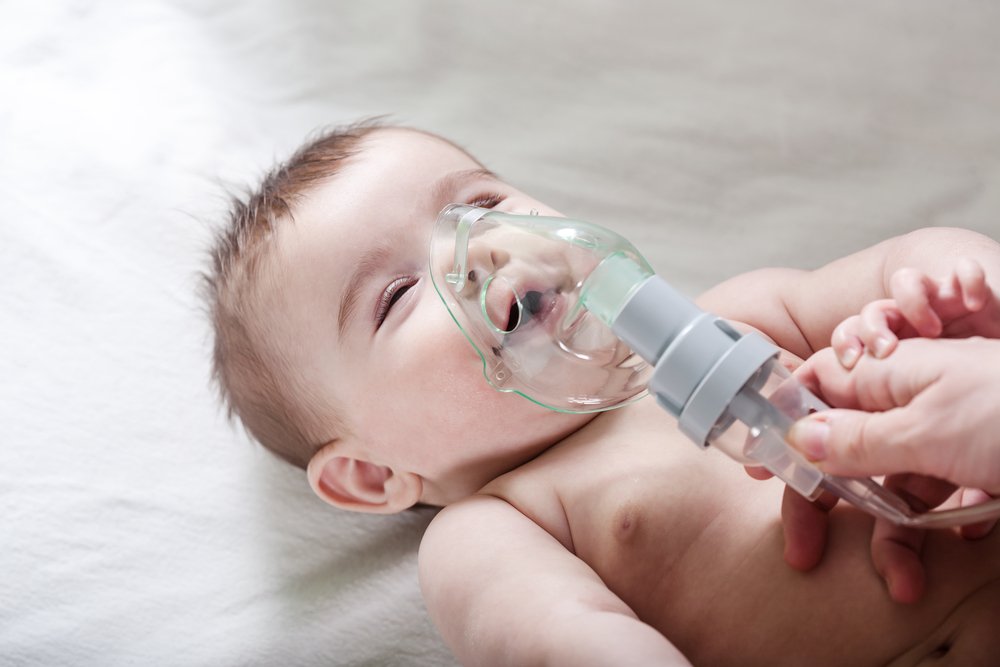
Муковисцидоз – наследственное заболевание, которое встречается у одного из 7 000 – 10 000 новорожденных. Оно связано с повреждением (мутацией) особого гена. Для того чтобы заболевание проявилось, ребенок должен получить две мутации (по одной от каждого из родителей). Родители больного ребенка всегда здоровы, но являются носителями патологического гена. До рождения больного ребенка родители не знают о своем носительстве. Риск рождения больного ребенка в браке двоих таких носителей составляет 25% при каждой беременности. Все остальные родственники чаще всего здоровы.
Как проявляется заболевание?
Повреждение гена приводит к нарушению транспорта солей через мембраны клеток, в результате чего страдает работа желез в пищеварительной и дыхательной системе, потовых желез. Их секрет становится вязким, он с трудом проходит по протокам и вызывает нарушение работы органов дыхания и пищеварения, пот ребенка становится соленым (это и определяет потовый тест). Большинство детей, имеющих заболевание, выглядят здоровыми при рождении. Но уже в первые месяцы жизни появляются затяжной кашель, частые бронхиты и пневмонии, нарушения стула, вздутие живота и боли в животе, задержка физического развития.
Небольшая часть детей с муковисцидозом при рождении имеют тяжелое состояние, связанное с непроходимостью кишечника (так называемый «мекониальный илеус»). Оно требует неотложной хирургической помощи.
Какое существует лечение заболевания?
Для лечения используются ферменты, антибиотики, препараты, способствующие отхождению мокроты, физиотерапия. Раннее начало лечения помогает улучшить состояние здоровья, хотя и не предотвращает прогрессирования болезни.
Кто будет наблюдать ребенка?
После постановки диагноза муковисцидоз малышу потребуется консультация врача-пульмонолога - специалиста по муковисцидозу в «Центре муковисцидоза» ГБУЗ «Детская краевая клиническая больница» министерства здравоохранения Краснодарского края. Врач индивидуально определит, какими специалистами и как часто должен наблюдаться ребенок. Семье также будет предложено пройти консультацию врача-генетика и генетическую диагностику для определения мутаций, вызвавших заболевание. Это обследование важно как для ребенка, так и для других членов семьи.
ЧТО ТАКОЕ ГАЛАКТОЗЕМИЯ?
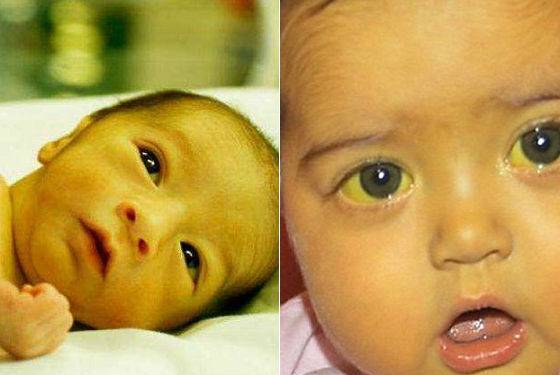
Галактоземия – наследственное заболевание обмена веществ, которое встречается у одного из 30 000 – 100 000 новорожденных. Это достаточно редкое заболевание. Галактоземия возникает, если в организме отсутствует способность усваивать сахар, содержащийся в молоке. Это происходит из-за нарушения (мутации) в структуре гена, который отвечает за синтез фермента, расщепляющего галактозу, одного из простых сахаров. Больной ребенок рождается, если он получает два мутантных гена (по одному от каждого из родителей). Родители больного ребенка всегда здоровы, но являются носителями патологического гена. До рождения больного ребенка родители не знают о своем носительстве. Риск рождения больного ребенка в браке двоих таких носителей составляет 25% при каждой беременности. Все остальные родственники чаще всего здоровы.
Как проявляется заболевание?
Первые признаки болезни могут появиться уже на первой неделе жизни новорожденного, и они связаны с нарушением работы печени. У новорожденного наблюдается желтуха и низкое содержание сахара в крови. Затем могут возникнуть различные симптомы поражения нервной системы в виде вялости или судорог, а также рвота, понос и другие нарушения со стороны желудочно-кишечного тракта. У больных галактоземией появляется поражение глаз, чаще всего в виде катаракты, нередко развиваются цирроз печени и умственная отсталость. Всех этих тяжелых клинических проявлений галактоземии можно избежать, если вовремя начать лечить ребенка. Если лечение начато рано, то клинические симптомы галактоземии у ребенка не проявляются, и он может расти здоровым, практически не отличаясь от сверстников.
Какое существует лечение заболевания?
Лечение заключается в исключении пищевых продуктов, содержащих галактозу, прежде всего грудного молока и других молочных смесей. Они могут быть заменены специальными смесями, приготовленными на основе сои, которые не содержат галактозу. Успех лечения во многом определяется тем, насколько родители больного ребенка осознали важность диетотерапии и насколько строго они ее выполняют.
Кто будет наблюдать ребенка в случае подтверждения диагноза?
О заболевании семье расскажет врач-генетик во время первого визита семьи в медико-генетическую консультацию. Основным врачом, наблюдающим больного ребенка, будет врач-диетолог, это может быть педиатр или врачгенетик, имеющий опыт в диетотерапии, который научит родителей расчету диеты для ребенка. У ребенка будет постоянно контролироваться содержание галактозы в крови. В зависимости от значений лабораторных показателей, будет корректироваться состав тех продуктов, которые, с одной стороны, не будут повышать уровень галактозы, а с другой, обеспечивать нормальный рост и развитие ребенка.
ЧТО ТАКОЕ СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ?
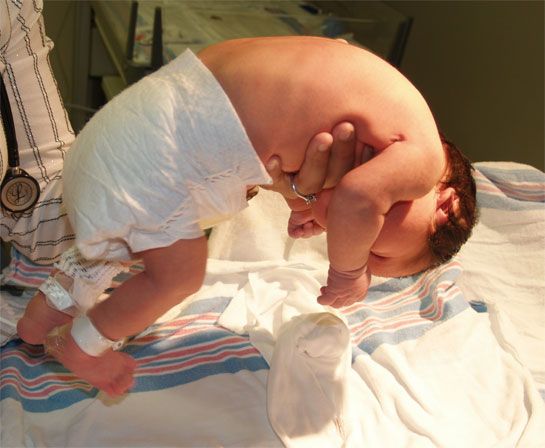
Спинальная мышечная атрофия – наследственное заболевание нервной системы, при котором из-за генетического нарушения происходит прогрессирующее поражение клеток нервной системы, отвечающих за работу скелетных мышц. Спинальная мышечная атрофия встречается у одного человека из 5000-10000. Больной ребенок рождается у двух здоровых родителей. При этом оба родителя являются носителями одной измененной копии гена SMN1. Риск рождения ребенка со спинальной мышечной атрофией в таком браке составляет 25% при каждой беременности. До рождения больного ребенка, либо до проведения специальных генетических тестов на определение носительства родители не могут знать о риске рождения ребенка со спинальной мышечной атрофией.
Как проявляется заболевание?
Возраст появления первых симптомов варьируется от первых дней жизни до взрослого возраста в зависимости от типа спинальной мышечной атрофии. Основными симптомами являются мышечная слабость в руках, ногах и туловище, задержка двигательного развития, либо постепенная потеря имеющихся двигательных навыков. При самом тяжелом I типе спинальной мышечной атрофии быстро развиваются нарушения дыхания и глотания. Чем раньше появляются симптомы, тем они тяжелее и быстрее прогрессируют. Психическое развитие людей со спинальной мышечной атрофией не нарушено.
Какое существует лечение заболевания?
На сегодняшний день существуют несколько генотерапевтических препаратов, влияющих непосредственно на причину заболевания. Эффективность терапии напрямую зависит от возраста постановки диагноза спинальной мышечной атрофии, и наилучшие результаты достигаются, если лечение начато до появления первых симптомов (досимптоматическая стадия болезни).
О заболевании расскажет врач-генетик во время первого визита семьи в медико-генетическую консультацию. При подтверждении диагноза ребенок будет направлен на консультацию к врачу-неврологу в детский диагностический центр ГБУЗ «Детская краевая клиническая больница» министерства здравоохранения Краснодарского края. Комплексное клиническое обследование будет проведено в психоневрологическом отделении ГБУЗ «Детская краевая клиническая больница» министерства здравоохранения Краснодарского края. По результатам данного обследования будет выбрано наиболее подходящее лечение. В дальнейшем ребенок будет наблюдаться врачом-неврологом и другими специалистами.
Врач–генетик назначит необходимые молекулярно-генетические обследования родителям и, в ряде случаев, другим близким родственникам, даст рекомендации по планированию последующих беременностей в семье.
ЧТО ТАКОЕ ПЕРВИЧНЫЙ ИММУНОДЕФИЦИТ?
Первичный иммунодефицит – это группа наследственных заболеваний иммунной системы. Существует целый ряд генов, мутации в которых приводят к нарушению работы иммунной системы. В некоторых случаях кто-то из родителей или близких родственников больного ребенка может иметь аналогичное заболевание, но очень часто ребенок с первичным иммунодефицитом рождается у абсолютно здоровых родителей. Каждое отдельное заболевание встречается редко, но суммарная частота всех заболеваний этой группы достигает 1 на 10000 новорожденных.
Как проявляется заболевание?
Возраст появления первых симптомов варьирует от первых дней жизни до взрослого возраста, однако большинство тяжелых форм первичного иммунодефицита проявляются в первые недели или месяцы жизни частыми и/или тяжело протекающими инфекциями различных органов. В раннем возрасте могут развиваться аутоиммунные болезни, например, сахарный диабет, а также злокачественные опухоли. В ряде случаев болезнь протекает стремительно и приводит к жизнеугрожающему состоянию у совсем маленьких детей.
Какое существует лечение заболевания?
Для лечения используются препараты, которые замещают нарушенную функцию иммунитета. Такое лечение может быть пожизненным, но во многих случаях оно приводит к полной компенсации и нормальному развитию ребенка. В тяжелых случаях необходима трансплантация донорских гемопоэтических клеток.
Кто будет наблюдать ребенка в случае подтверждения диагноза?
Все дети, попавшие в группу риска первичного иммунодефицита по результатам расширенного неонатального скрининга, направляются на консультацию и подтверждающую диагностику к врачу-иммунологу детского диагностического центра ГБУЗ «Детская краевая клиническая больница» министерства здравоохранения Краснодарского края. При подтверждении диагноза ребенок будет наблюдаться врачом-иммунологом ГБУЗ «Детская краевая клиническая больница» министерства здравоохранения Краснодарского края. Врач–генетик назначит необходимые молекулярно-генетические обследования родителям и, в ряде случаев, другим близким родственникам, даст рекомендации по планированию последующих беременностей в семье.
ЧТО ТАКОЕ НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА АМИНОКИСЛОТ, ОРГАНИЧЕСКИХ КИСЛОТ И ЖИРНЫХ КИСЛОТ?

Наследственные болезни обмена аминокислот, органических кислот и жирных кислот - большая группа редких генетических заболеваний, при которых из-за мутаций в определенных генах нарушается работа того или иного фермента в важнейших биохимических реакциях организма, что приводит к накоплению токсических веществ, нехватке энергии и питательных веществ. В результате страдают все органы и системы, но прежде всего – нервная система, сердце, печень. Частота каждого заболевания низкая – один больной на 10 000 – 500 000 новорожденных детей, но суммарная частота 29 заболеваний, включенных в расширенный неонатальный скрининг, существенно выше. У большинства болезней этой группы аутосомно-рецессивный тип наследования, при котором больной ребенок рождается у здоровых родителей – носителей мутации в скрытом виде. Также здоровы и все старшие родственники со стороны мамы и папы. Однако в семье данных родителей существует высокий риск (25%) повторного рождения ребенка с аналогичным заболеванием обмена веществ.
Как проявляются эти заболевания?
Заболевания этой группы могут начаться в любом возрасте, но чаще всего – в первые дни или недели жизни. Важной особенностью является их жизнеугрожающий характер. Зачастую заболевание развивается стремительно и в течение нескольких часов может привести к необратимым изменениям в головном мозге и даже - к внезапной смерти ребенка (так называемый «метаболический криз»). В ряде случаев первым проявлениям болезни предшествует «светлый промежуток» продолжительностью до нескольких месяцев или даже лет, когда ребенок выглядит абсолютно здоровым, и без проведения специальных лабораторных исследований невозможно заподозрить наличие у него такого опасного заболевания. Если лечение не назначено вовремя, до появления первых симптомов, то часть заболеваний этой группы с высокой вероятностью приводят к смерти ребенка при развитии метаболического криза, при других же формах развивается тяжелая хроническая прогрессирующая патология нервной системы, задержка развития и поражение всех органов.
Какое существует лечение заболеваний этой группы?
При разных болезнях из этой группы лечение будет различаться, но в большинстве случаев применяется специальная диета с ограничением определенных продуктов питания, назначаются специализированные продукты лечебного питания, а также препараты, влияющие на нарушенные звенья обмена веществ и связывающие избыток токсических веществ в организме. Эффективность терапии напрямую зависит от возраста постановки диагноза наследственной болезни обмена, и наилучшие результаты достигаются, если лечение начато до появления первых симптомов (досимптоматическая стадия болезни).
Кто будет наблюдать ребенка в случае подтверждения диагноза?
О заболевании расскажет врач-генетик во время первого визита семьи в центр. При серьезном подозрении на НБО в ряде случаев ребенку будет рекомендована неотложная госпитализация для проведения углубленного клинического обследования, мониторинга его состояния и начала терапии. В дальнейшем, при подтверждении диагноза, ребенок будет наблюдаться педиатром соответствующего профиля ГБУЗ «Детская краевая клиническая больница» министерства здравоохранения Краснодарского края совместно с врачом-генетиком медико-генетической консультации, кроме того, ребенка будут наблюдать другие специалисты в поликлинике по месту жительства. Врач– генетик назначит необходимые молекулярно-генетические обследования родителям и, в ряде случаев, другим близким родственникам, даст рекомендации по планированию последующих беременностей в семье.
























